>
> Protein Representation Learning by Geometric Structure Pretraining

The study of structural biochemistry is based on the axiom that “structure determines function”. A corollary of that axiom is that, for proteins, “function is independent of sequence, given structure”. That is, knowing the sequence of a protein is helpful for determining function insofar as it informs us about the structure of the protein. Many recent works including ESM-1b and ProtBERT-BFD have attempted to predict function from sequence. But in a recent preprint, Zhang et. al. achieved state-of-the-art accuracy by predicting function from structure, using self-supervised pre-training and the GearNet architecture.
Self-supervised pre-training “warms-up” a model to a prediction task. It involves training on auxiliary objectives that help the model learn better representations of the data. Labels are derived from the data itself, rather than being manually annotated, hence the term “self-supervised”. Many of the aforementioned sequence-to-function networks use self-supervised pre-training to learn better sequence representations, aiding them in achieving high-accuracy function prediction. In the case of GearNet, the model uses auxiliary structure-based prediction tasks which make it easier to learn the downstream function prediction objective.
Dataset
Until recently, protein structure databases have been limited to experimental structures, limiting the effectiveness of structure-based representation learning. However, AlphaFold recently made possible the creation of massive datasets of predicted protein structures. The 800k AlphaFold-predicted structures in the combined AlphaFold Proteome and AlphaFold Swiss-Prot datasets augment the 182k experimental structures in the Protein Data Bank to bring the available structure datasets up to a scale suitable for massive, structure-based pre-training.
From the protein structure files, the authors generate a graph representation G of each protein. Nodes in the graph represent the alpha carbon of each residue, with a one-hot vector indicating residue identity. Edges in the graph are relational, meaning that there are multiple types of edges (or “relations”). Some edge types indicate proximity in sequence, while other edge types indicate spatial proximity.
Besides the original graph, GearNet also creates a “supergraph” G’ that treats relational edges as nodes and creates new directed edges that connect incoming edges to outgoing edges.
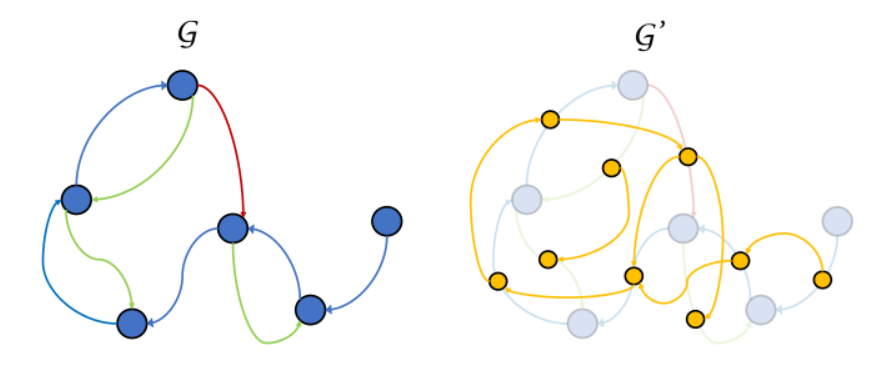
The original graph G and the supergraph G’. Different colors represent different relational edge types.
Model Architecture
GearNet is a “Geometry-Aware Relational Graph Neural Network”. It takes vector representations of nodes and edges as inputs, conducts updates to the node features and edge features, and predicts updated node features.
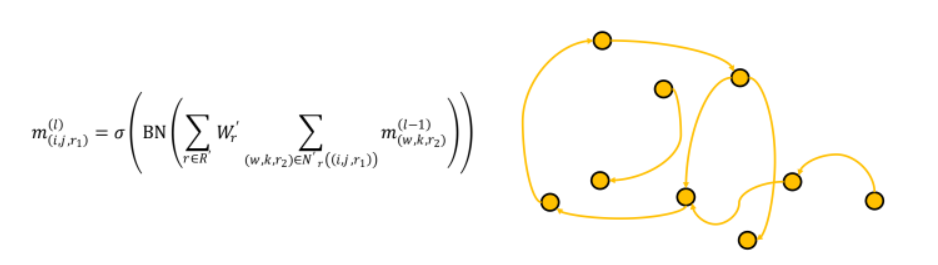
GearNet conducts two types of updates. The first update operates on G’, and updates edge features based on edges in its neighborhood.
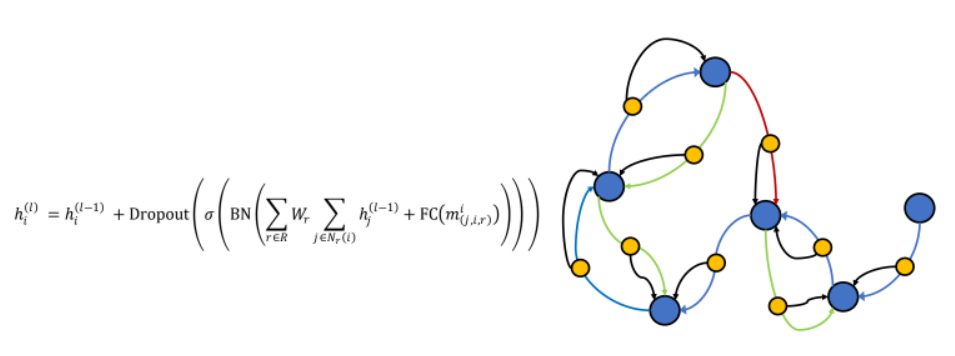
The second update operates on G, and updates node features based on nodes and edges it its neighborhood.
Self-Supervised Objectives
The model is pre-trained on five self-supervised tasks. The first is called “Multiview Contrastive Learning”. In this method, the model is given two different crops of each protein in a batch. The contrastive objective encourages the model to learn similar representations between crops from the same protein, while discouraging it from producing representations similar to crops from other proteins in the batch.
The other four self-supervised techniques are various types of missing information prediction problems. They involve predicting residue type, predicting distance between nodes, predicting angles between edges, and predicting dihedral angles.
Experiments and Results
Once the network is pre-trained, it is “fine-tuned” by adding a “projection” fully connected layer and training the entire model for the specific prediction task. The authors evaluated the network on Enzyme Commission classification, Gene ontology classification, fold classification, and reaction classification [very similar to Enzyme Commission classification]. Their model achieves state-of-the-art performance in almost all benchmarks, while training on several orders of magnitude less data than sequence-based methods. In the best case, for fold classification, GearNET achieves a 7 raw percentage point increase over the next best model.
Why does this work?
By learning these auxiliary tasks [e.g. predicting the identity of a masked residue, predicting whether two sub-proteins come from the same protein], the neural network needs to use structural information to infer other structural information. This is similar to the task of using structural information to predict function. So after self-supervised pre-training, the model can converge quickly to an optimum for the function prediction task.
Takeaways:
- Self-supervised representation learning helps for downstream function prediction tasks.
- Representation learning on structure seems to be more useful and data-efficient than representation learning on sequence.
Structure-based methods also lend themselves to structure-based interpretability, helping to identify the structural reason for function prediction.