>
> Design of Protein Binding Proteins from Target Structure Alone

How do you design a protein that binds to another protein given only the target protein structure?
Until recently, you could use Rosetta to manually craft a protein using expert heuristics. However, this process is laborious, expensive, and does not generalize. Researchers at the Institute for Protein Design recently published a groundbreaking work outlining a systematic process for the design of protein binders, which generalizes to a diverse set of 12 target proteins.
A Rock Climbing Analogy
The authors introduce their method with an analogy. Past approaches have identified the most promising binding “hotspots” on the target protein surface, and designed binders to maximize those hotspots. The authors compare this approach to tackling a climbing route by using only the very best holds. They describe their new approach as one that considers all possible climbing routes, even those which include poor holds. The idea is to take advantage of the accumulation of small interactions to design a strong binder.

The Process
The process outlined in the paper consists of eight steps. The first four steps involve screening a large protein scaffold library, which they call “Global Search”. The last four steps, which they call “Focused Search”, involve refinement of existing structures.
- Generation of a Rotamer Interacting Field (RIF)
In this step, billions of free amino acids are docked onto the target binding surface. Each docked amino acid is assigned a target interaction energy. - Scaffold-RIF docking
Next, a library of protein scaffolds is docked against the RIF. The interaction energy for each scaffold is approximated by taking the scaffold’s interface residues, finding their nearest amino acid match in the RIF, then summing the target interaction energies of the individual amino acids. This saves a huge amount of computation because it only computes the interaction energy once per docked amino acid, then reuses that energy for every scaffold that matches that amino acid. - Interface Sequence Design
Once promising scaffolds are obtained, the residues are modified to maximize shape and chemical complementarity with the target and to avoid buried polar atoms. - Interface Motif Extraction
From a set of optimized scaffolds, they extract all secondary structure motifs in good contact with the target protein. - Clustering and Selection of Privileged Motifs
These motifs are clustered, and a representative motif from each cluster is selected as a “privileged motif” (about 2000 privileged motifs per target). This is similar to step 1, but now with motifs instead of free amino acids. - Modification of Scaffolds to Incorporate Motifs
Next, the original scaffold library is modified to incorporate the privileged motifs. Each scaffold is superimposed on the motifs (similar to Step 2, but with motifs instead of single amino acids), and privileged motifs are transferred to the scaffold. - Interface Sequence Design
Step 3 is repeated with the new scaffolds which have been modified to incorporate the privileged motifs.
Selection of Best Designs
Based on protein interface metrics and stability of the folded binding protein, the best binders are selected for experimental validation.
One note: designs underwent one step of further optimization. After selecting the best designs, the authors used site saturation mutagenesis (SSM) and found that this technique led to increased binding affinity in almost all cases.
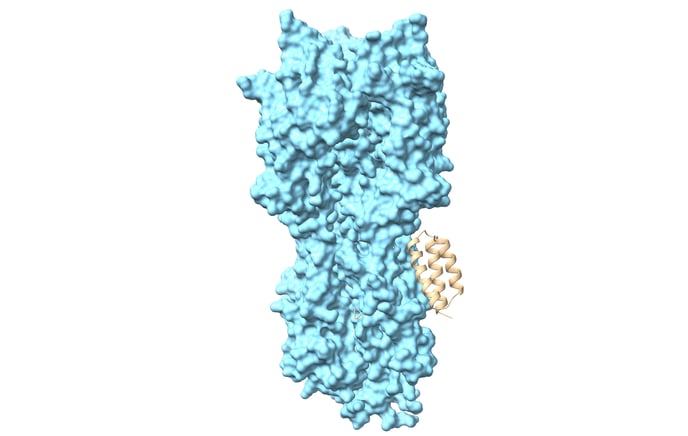
Caption: Influenza A H3 hemagglutinin with designed binder.
Experiments
This process was tested across a range of proteins. The authors considered two classes of target proteins: signaling proteins, for which binders can be used as probes of biological mechanism or as therapeutics; and pathogenic proteins, against which binders can be applied therapeutically. They also assessed a range of binding sites, from sites that overlap with known protein-protein interfaces, to sites with zero overlap with known binding regions.
Results
For all 12 protein targets, this process found protein-protein binders with binding affinity 990nM or better, with the best binders reaching 300pM binding affinity. This was achieved while maintaining high specificity to the target protein. Binding affinity and structure were verified via biolayer interferometry and X-ray crystallography.
Conclusions
In a time when deep learning-based approaches to protein design have generated much excitement, this is a general approach to protein design that demonstrates the validity of traditional computational methods.